Introduction
Materials
Methods
Choosing the right primer combination
Template preparation
AFLP pre-amplification
AFLP amplification
Comments
Introduction
The AFLP technique, originally known as selective restriction fragment amplification (SRFA) (Zabeau and Vos 1993), produces highly complex DNA profiles by arbitrary amplification of restriction fragments ligated to double-stranded adaptors with hemi-specific primers harboring adaptor-complementary 5' termini (Vos et al. 1995). The technique has been widely used in the construction of genetic maps containing high densities of DNA marker loci. The AFLP protocol amplifies restriction fragments obtained by endonuclease digestion of target DNA using "universal" AFLP primers complementary to the restriction site and adapter sequence. However, not all restriction fragments are amplified because AFLP primers also contain selective nucleotides at the 3' termini that extend into the amplified restriction fragments. These arbitrary terminal sequences result in the amplification of only a small subset of possible restriction fragments. The number of amplified fragments (generally kept around 50-100) can therefore be "tailored" by extending the number of arbitrary nucleotides added to the primer termini. Alternatively, the use of endonuclease combinations that vary in their restriction frequency can also be used to tune the number of amplicons. Generally, the abundant restriction fragments produced from complex genomes require of AFLP primers with longer selective regions. Conversely, analysis of small genomes require of only few arbitrary nucleotides added at the primer 3' termini. The resulting AFLP fingerprints are usually a rich source of DNA polymorphisms that can be used in mapping and general fingerprinting endeavors.
The AFLP protocol can be devided in the following steps: (1) DNA digestion with two different restriction enzymes (generally a rare and a frequent cutter), (2) ligation of double-stranded adapters to the ends of the restriction fragments, (3) optional DNA pre-amplification of ligated product directed by primers complementary to adapter and restriction site sequences, and (4) DNA amplification of subsets of restriction fragments using selective AFLP primers and labelling of amplified products. Amplification of very small "genomes" (plasmids, cosmids, BACs) requires of primers with no selective nucleotides. AFLP fingerprinting of bacteria and fungi generally requires primers with 2 selective bases. Complex genomes require the use of more than 2 selective bases in one or both primers. In the case of complex genomes it is recomended to carry the amplification in two consecutive steps (preamplification and selective amplification) to increase specificity and the amount of initial template. The AFLP fragments are usually detected by labeling one of the two AFLP primers. For example, radioactively labeled primers can be obtained by phosphorylating the 5' ends with g-33P-ATP and polynucleotide kinase. Do not label the two primers if the generation of doublets resulting from the different mobility of complementary strands in sequencing gels wants to be avoided. Finally, the labelled reaction products are separated by electrophoresis using denaturing polyacrylamide gels and exposed to X-ray films to visualize the AFLP fingerprints. Return
Materials
1. Equipment: thermocycler, sequencing gel rig, high voltage power supply
2. Disposables: PCR tubes, X-ray film
3. Genomic DNA. For genomes smaller or larger than 500 Mb use 50 ng or 100 ng DNA for template preparations, respectively. Determine DNA concentrations by measuring OD260, and confirm the measurement and the integrity of DNA by electrophoresing the sample together with a series of phage l DNA dilutions ranging from 50 ng to 500 ng in agarose gels.
4. AFLP primers (50 ng/µL). Primers are named "+0" when having no selective bases, "+1" when having a single selective base, "+2" for having two selective bases, and so on.
EcoRI-primer +0: 5'-GACTGCGTACCAATTC-3'
EcoRI-primer +1: 5'-GACTGCGTACCAATTCA-3'
EcoRI-primers +2: 5'-GACTGCGTACCAATTCAN-3'
EcoRI-primers +3: 5'-GACTGCGTACCAATTCANN-3'
PstI-primer +0: 5'-GACTGCGTACATGCAG-3'
PstI-primer +1: 5'-GACTGCGTACATGCAGA-3'
PstI-primers +3: 5'-GACTGCGTACATCGAGANN-3'
MseI-primer +1: 5'-GATGAGTCCTGAGTAAC-3'
MseI-primers +2: 5'-GATGAGTCCTGAGTAACN-3'
MseI-primers +3: 5'-GATGAGTCCTGAGTAACNN-3'
TaqI-primer +0: 5'-GATGAGTCCTGAGCGAA-3'
TaqI-primers +3: 5'-GATGAGTCCTGAGCGAANN-3'
5. Corresponding adaptors (5 or 50 pmol/µL):
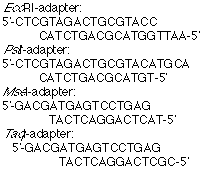
6. Double-distilled water.
7. Buffers: 1 M Tris.HAc pH 7.5; 1 M Tris.HCl pH 8.0 and pH 8.3.
8. Magnesium: 0.1 mM MgCl2.
9. TE (10x): 100 mM Tris.HCl, 10 mM EDTA pH 8.0.
10. 100 mM DTT.
11. 5 mM of dNTPs
12. gamma-32P-ATP (~3000 Ci/mmol) or gamma-33P-ATP (~2000 Ci/mmol).
13. 10 mM ATP.
14. T4-buffer (10x): 250 mM Tris.HCl pH 7.5, 100 mM MgCl2, 50 mM DTT, 5 mM spermidine
15. Restriction-ligation buffer (5x): 50 mM Tris-HAc, 50 mM MgCl2, 250 mM KAc, 25 mM DTT, 250 ng/µL, pH 7.5.
16. Restriction endonucleases: EcoRI, PstI, MseI, TaqI (New England Biolabs).
17. Enzymes: T4 DNA ligase, T4 polynucleotide kinase, Taq DNA polymerase
18. PCR buffer (10x): 100 mM Tris.HCl pH 8.3, 15 mM MgCl2, 500 mM KCl.
19. Molecular weight standards
20. General reagents for polyacrylamide gel electrophoresis. Return
Methods
Choosing the right primer combination
Depending on the size of the genome to be analyzed a different set of primers will have to be used. High complexity genomes require of a pre-amplification step. The following table shows primer combinations required for the amplification of different genomes. Note that numbers depict the number of selective nucleotides at the 3' terminus of the individual primers.
| Genome | Endonucleases | Pre-amplification | Amplification |
| Cosmids, BACs, PACs, YACs (0.01-1 Mb) | EcoRI-MseI | – | 0-0 or 0-1 |
| Microbes (1-5 Mb) | EcoRI-MseI | – | 1-1 |
| Microbes (5-20 Mb) | EcoRI-MseI | – | 1-2 |
| Fungi (20-100 Mb) | EcoRI-MseI | – | 2-2 |
| Plants, invertebrates (100-500 Mb) | EcoRI-MseI | 0-1 | 2-3 |
| Plants (500-5000 Mb) | EcoRI-MseI | 1-1 | 3-3 |
| Plants (more than 5000 Mb) | PstI-MseI | 1-1 | 3-3 |
| Vertebrates (3000 Mb) | EcoRI-TaqI | 1-1 | 3-3 |
Note that depending on the DNA to be analyzed individual endonuclease and primer combinations and required with consequent change in the protocol.
Template preparation
1. Adapters: Prepare double-stranded (ds) adapters by mixing individual synthetic oligonucleotides. No denaturing-renaturation step is required. EcoRI, MseI, PstI, and TaqI adapters have double-stranded regions of 14, 12, 14 and 12 base pairs, respectively. Mix 1500 pmoles of each oligonucleotides to produce 5 pmol/µL solutions. Generally, use 8.5 µg, 10.5 µg, 8 µg or 8 µg of the top strand oligonucleotide with 9.0 µg, 7 µg, 7 µg or 7 µg of the bottom strand oligonucleotidein 300 µL of water for the EcoRI, PstI, MseI and TaqI adapter, respectively.
2. Digestion of DNA: Digest genomic DNA (50-100 ng) with restriction endonucleases in a 40 µL reaction containing DNA, 8 µL 5x RL buffer, 5 units EcoRI and 2 units MseI. Mix well and incubate for 2 h at 37°C. For genomes larger than 5000 Mb use 5 units PstI and 2 units MseI. For mammalian and vertebrate genomes use 5 units TaqI and 5 units EcoRI in subsequent incubation steps at 65°C and 37°C.DNA preparations need to be of sufficient quality to allow complete digestion, since this step is crucial for the production of good quality AFLP fingerprints. Often, contaminating agents can interfere with digestion.
3. Adaptor ligation: Ligate adaptors to the digested genomic DNA by adding 1 µL EcoRI adapter (5 pmol), 1 µL MseI adapter (50 pmol), 1 µL PstI adapter (5 pmol) or 1 µL TaqI adapter (50 pmol), 1 µL 10 mM ATP, 2 µL 5¥ RL buffer, 1 unit T4 DNA ligase and 5 µL water to the digestion mix. Incubate another 2 h at 37°C. Overall, DNA is incubated for a total of 4 h with endonucleases, the last 2 h in the presence of T4 DNA ligase and oligonucleotide adapters. Avoid longer incubation periods because of possible "star" activity of EcoRI that gives reduced cleavage specificity and aberrant AFLP fingerprints. When using TaqI, use high concentration of adapters; if not TaqI will not efficiently re-digest aberrant fragment-ligation products at 37°C.
4. Dilution: Dilute the ligation reaction mixture 10 times with TE (usually 10 µL in 100 µL) and use the diluted reaction mixture directly as template DNA for the AFLP reactions. Store diluted DNA at -20°C.
AFLP pre-amplification
1. Pre-amplification: Check above for the right combination of primers for your individual application. If genomic complexity is sufficiently low, AFLP preamplification is not required(see Table above); therefore, skip this preamplification section. Assemble the preamplification reaction (50 µL total volume) with following components: 5 µL ligated DNA, 1.5 µL EcoRI-primer +0 (75 ng), 1.5 µL MseI-primer +C (75 ng), 1.5 µL of the PstI one-base extension primer -A (75 ng), 1.5 µL MseI-primer +C (75 ng) and/or 1.5 µL TaqI-primer +A (75 ng), 2 µL 5 mM dNTPs, 0.2 µL Taq polymerase (1 unit), 5 µL 10x PCR-buffer and 34.8 µL water. Preamplify the mix for 20 cycles using the following regime: 30 s at 94°C; 60 s at 56°C; 60 s at 72°C. For mammalian genomes use 30 cycles. For genomes larger than 5000 Mb modify the amplification regime as follows: a first cycle of 30 s at 94°C, 60 s at 65°C, 60 s at 72°C, followed by 12 cycles with a stepwise decrease of the annealing temperature in each subsequent cycle by 0.7°C, and 23 cycles of 30 s at 94°C, 30 s at 56°C and 60 s at 72°C. In this step it is advisable to assemble one reaction mix containing primer and dNTPs and another containing the Taq polymerase and its buffer.
2. Dilution: Preamplification, 10 µL of the reaction is diluted with 190 µL of TE0.1 to 100 µL which is sufficient for 40 AFLP-reactions +2/+3. The diluted reaction mix and the rest of the preamplification reaction is stored at -20°C. If necessary new dilutions of the preamplification reactions may be made to give additional template for the AFLP reactions.
Important note: Since ligation uses non-phosphorylated adapters, only one strand of the adapter binds to the DNA with adapters ligated to opposite strands. The recessed 3' ends of the template DNA are filled-in by the Taq polymerase during heating to 94°C of the first PCR-cycle. Therefore, if template DNA is denatured prior to the start of the amplification, amplification will be prevented.
AFLP amplification
1. Preparation of labelling mix: Label primers for selective AFLP amplification by phosphorylating the 5' end of the primers with gamma-32P-ATP or gamma-33P-ATP and polynucleotide kinase. Check above for the right primer combination to use in this step. Only one of the two primers of the AFLP reaction should be labelled (e.g., the EcoRI-primer). When possible use the more expensive 33P-labelled primers because they give better product resolution in polyacrylamide gels, and are less prone to degradation due to autoradiolysis. Prepare the following primer labelling mixes (40 µL) for 100 AFLP reactions. either 20 µL gamma-32P-ATP (~3,000 Ci/mmol) or 10 µl gamma- 33P-ATP (~2,000 Ci/mmol), 5 µL 10xT4-buffer, 2 µL T4-kinase (10 units/µL) and water to 40 µL.
2. Primer labelling: Add 10 µl of primer (either EcoRI- or PstI-primers at 50 ng/µL) to 40 µL labelling mix and incubate 60 min at 37°C, followed by incubation at 70°C for 10 min for the inactivation of the kinase. This gives a labelled primer with a concentration of 10 ng/µL.
3. Prepare AFLP reaction mixes: Prepare reaction mixes for a minimum of 10 reactions. Working with AFLP reaction mixes is important for the reliability and reproducibility of AFLP reactions and because it facilitates reaction assembly. Primer and dNTPs mix (50 µL): 5 µL labelled primer (10 ng/µL), 6 µL unlabelled primer (50 ng/µL), 8 µL 5 mM dNTPs and 31 µL water. Taq polymerase mix (100 µL): 20 µL 10x PCR-buffer, 0.8 µL Taq polymerase (4 units) and 79.2 µL water.
4. AFLP amplification: Assemble the reaction by adding 5 µL of the primers and dNTPs mix and 10 µL of the Taq polymerase mix to 5 µL of pre-amplified ligated DNA. The template DNA should be pipetted first followed by the two mixes. The reagents should be mixed by tapping the base of the tubes on the bench. Pipetting mixes is essential for the rapid start of the AFLP reactions that are assembled at room temperature (to avoid loss of AFLP fingerprint quality). Tubes are amplified in a thermocycler with the following cycle regime a first cycle of 30 s at 94°C, 30 s at 65°C and 60 s at72°C, followed by 12 cycles with a stepwise decrease of the annealing temperature in each subsequent cycle by 0.7°C, and 23 cycles of 30 s at 94°C, 30 s at 56°C and 60 s at 72°C. The reaction is started at a high annealing temperature to obtain optimal primer selectivity. In the following steps the annealing temperature is lowered gradually to a temperature for optimal primer annealing.
Polyacrylamide gel electrophoresis of AFLP products
1. General: Amplification products are analyzed on 4.5% denaturing polyacrylamide sequencing gels (see the Separation section for more details). Treat back glass plate of the gels with 2 mL of repel silane, and the front plate with 10 mL of bind silane solution (30 µL acetic acid and 30 µL bind silane in 10 mL ethanol, freshly made immediately before use). The gels should be cast at least 2 h before use and should be prerun for 0.5 h just before loading the samples Perform pre-running and running electrophoretic steps at 110 W. Use TBE (1x) as running buffer.
2. Sample loading: Mix AFLP reaction products with an equal volume (20 µL) of loading dye. Heat the samples for 3 min at 90°C, and then quickly cool on ice. Rinse the the gel wells with running buffer and push carefully two 24-well sharktooth combs about 0.5 mm into the gel surface to create the gel slots. Rinse the gel slots formed in this way with TBE and load 2 µL of each sample per well.
3. Post-electrophoretic procedures: Disassemble the gel cassette and remove the front glass plate with the silane-attached gel to the front. Fix the gel by soaking in 10% acetic acid for 30 min and dry it subsequently at room temperature in a fume hood for 10-20 h. Autoradiographic exposure of the 32P-gels to standard X-ray film overnight, without intensifying screens. Exposure of the 33P-gels takes 2 to 3 d in order to generate similar band intensities. Return
Comments
AFLP can be used in the fingerprinting for genomic DNA of varying origins and complexities. The amplification reaction is stringent, versatile and robust, and appears to be quantitative. While AFLP is capable of producing very complex fingerprints, it is a technique that requires DNA of reasonable quality and is more experimentally demanding than its arbitrarily amplified DNA (AAD) counterparts. The advantage of AFLP is its high multiplexity and therefore the possibility of generating high marker densities. In this respect, AFLP and DAF are comparable. One limitation of the AFLP technique is that fingerprints may share few common fragments when genome sequence homology is less than 90%. Therefore, AFLP cannot be used in comparative genomic analysis with hybridization-based probes or when comparing genomes that are ecolving rapidly such as those of some microbes (Janssen et al. 1996). Conversely, very homogeneous genomes may not be suitable for AFLP analysis. Other techniques that use fingerprint tailoring strategies (such as ASAP) may be used in this respect.
The molecular basis of AFLP sequence polymorphisms rests on the detection of single nucleotide changes within restriction sites or adjacent nucleotides used for AFLP primer annealing. In addition, polymorphisms can also result from deletions, insertions and rearrangements affecting the presence or restriction sites and/or adjacent sequences. Therefore, most AFLP markers will be mono-allelic with only few being di-allelic. Return

